

Products Of Acid Base Reaction
Chapter 1: Acid–Base Reactions
 As we will come across, organic reactions tin can be classified using a small set up of reaction types—the largest and most all-encompassing of which are those involving acid–base reactions. Agreement acrid–base reactions, therefore, provides a broadly useful conceptual framework within which to consider a wide range of organic reactions. Although information technology is probable that you have already been introduced to acid–base of operations reactions (especially if y'all used the Clue general chemistry curriculum[1]), we are going to review this class of reactions in guild to emphasize their general features. Our goal is that y'all learn how to recognize their office in a range of reaction mechanisms; understanding how and why acid–base reactions occur will give you to a ready of tools to sympathise phenomena as diverse equally why virtually drugs are usually administered every bit in their salt form (a conjugate acrid or base of operations), why biological systems are buffered to specific pH levels (and why unlike pH levels are establish in unlike cellular and organismic compartments), and why molecular oxygen (O2) transport systems require a metal ion complex (inside the proteins involved, e.g. myoglobin, hemoglobin, cytochromes). Equally we volition see, acid–base reactions are past far the most common types of reactions in biological systems.
As we will come across, organic reactions tin can be classified using a small set up of reaction types—the largest and most all-encompassing of which are those involving acid–base reactions. Agreement acrid–base reactions, therefore, provides a broadly useful conceptual framework within which to consider a wide range of organic reactions. Although information technology is probable that you have already been introduced to acid–base of operations reactions (especially if y'all used the Clue general chemistry curriculum[1]), we are going to review this class of reactions in guild to emphasize their general features. Our goal is that y'all learn how to recognize their office in a range of reaction mechanisms; understanding how and why acid–base reactions occur will give you to a ready of tools to sympathise phenomena as diverse equally why virtually drugs are usually administered every bit in their salt form (a conjugate acrid or base of operations), why biological systems are buffered to specific pH levels (and why unlike pH levels are establish in unlike cellular and organismic compartments), and why molecular oxygen (O2) transport systems require a metal ion complex (inside the proteins involved, e.g. myoglobin, hemoglobin, cytochromes). Equally we volition see, acid–base reactions are past far the most common types of reactions in biological systems.
A quick review of the models of acid–base reactions.
At that place are a number of ways to discuss acid–base reactions, depending on what aspects of the reaction we want to highlight. They range from the extremely simplified (and not useful) Arrhenius model, to the Brønsted–Lowry model that we use merely for reactions in which protons are transferred, and finally to the Lewis model, which can encompass any type of acid–base reaction.
Arrhenius: The Arrhenius acid–base model is probably the first acid–base model that you were introduced to in the form of your education. In this model, when an acrid dissolves in water information technology dissociates to release a hydrogen ion (H+); when a base of operations dissolves it releases a hydroxide ion (–OH).
Acid: HCl(grand) + H2O [latex]\rightarrow[/latex] H+ (aq) + Cl– (aq) (sometimes written as or HCl(aq))
Base: NaOH(s) + HtwoO [latex]\rightarrow[/latex] Na+(aq) + –OH(aq)
Acid–Base Reaction: HCl (aq) + NaOH(aq) [latex]\rightarrow[/latex] NaCl(aq) + H2O (l)[2]
Although simple, the Arrhenius model is not especially useful when it comes to understanding the reactions considered in organic chemistry. This of course raises the obvious question: and then why are we mentioning it? The answer is two fold: i) considering yous might well vaguely remember it as a description of acid–base behaviors and ii) then that we can consider why information technology is not useful and why y'all should not apply it. The Arrhenius acrid–base of operations model applies just when h2o is the solvent—as nosotros will see many organic reactions exercise non occur in water. The Arrhenius model as well, falsely implies that there are free protons (H+) roaming effectually in h2o and it restricts bases to those substances that release a hydroxide ion. Finally, it implies that an acid tin can exist independent of a base—and vice versa, which doesn't make a slap-up deal of sense.
Brønsted–Lowry: The Brønsted–Lowry model is a much more useful and flexible model for considering acid base reactions. In this model an acid is a proton (H+) donor and a base of operations is a proton acceptor. In the Brønsted–Lowry model you cannot have an acrid without a base, and vice versa; the acid has to donate its H+ to something (the base), and similarly the base has to accept it. The H+ doesn't merely "drop off"—it is transferred.[3] In the case of reactions that occur within aqueous solution, the H+ is transferred to a water molecule to grade H3O+. Consider, as an case, HCl; in aqueous solution HCl transfer a H+ group to a h2o molecule. The products are H3O+ (the conjugate acid of h2o) and Cl–, the conjugate base of HCl.
| HCl(thousand) + | H2O(fifty) | ⇆ | H3O+ (aq) | + | Cl–(aq) |
| acid | base | conjugate acid | conjugate base | ||
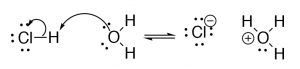
The central point here is that the H+ is transferred from i molecule to the other—information technology doesn't drop off and then reattach.
The flexibility of the Brønsted–Lowry model lies in the fact that the base does not necessarily have to be h2o. For example, if we wait at the reaction of hydrogen chloride and ammonia (NH3), we run into that the proton transfer from acrid to base is analogous to the reaction in water.
| HCl | + | NH3 | ⇄ NH4 + + | Cl– |
| acrid | base of operations | conjugate acid | cohabit base of operations | |
In the Brønsted–Lowry model, as for all chemic reactions considered at the molecular level, there is the possibility for the reaction to contrary, which is denoted by the use of equilibrium arrows (⇄).
At the macroscopic level the extent to which the reaction proceeds (from reactants on the left to products on the correct) is adamant past a number of factors. That is, we demand more information to predict (or calculate) the concentrations of reactants and projects at equilibrium. This is information that also enables us to predict whether the reaction will proceed in the forward direction (to the right) or not and how the reaction might change if nosotros add or remove reactants (or products).
We can place a potentially acidic H + because it volition exist bonded to a more than electronegative atom; the issue is that the electron density in the bond will lie mainly with the more electronegative cantlet (e.g. O, Due north, or Cl). The outcome is that, for example, an H–O bail will exist weakened (require less energy to break); the H volition have a large partial positive charge on it, and will be strongly attracted to basic centers (as described in the adjacent department). Similarly, simple bases can be identified by the presence of an atom (within the molecule) that has a partial negative charge; this partial negative accuse arises considering the atom (the basic middle) is bonded to less electronegative atoms. Now nosotros add i further consideration, this base of operations eye atom also needs to be able to accept the incoming H+. In do, this means that a basic molecule will contain an atom that has a lone (non-bonding) pair of electrons that tin can class a bond to the H + .
The Brønsted–Lowry model is useful for acid–base reactions that involve proton transfer, merely even so, information technology is express to proton transfer reactions. We also note here that the solvent in which the proton transfer takes identify will have an effect on the reaction, and we will render to this idea later in the course. If nosotros extend the Brønsted model to other reactions where a base uses its alone electron pair to course a new bond with an electropositive eye, we can aggrandize the class of acid–base reactions even further. Which brings us to the next model of acid base chemical science: the Lewis model.
Lewis: The Lewis model allows us to describe exactly the same ready of reactions equally does the Brønsted–Lowry model, but from a different perspective, and it also allows us to expand on the model. In the Lewis model a base has the power to donate an electron pair to form a new bond with the acid that accepts this new bond, often but not ever with the concomitant breaking of a bond within the acid molecule. We use same rationale for why the reaction occurs between two oppositely charged centers, but from the perspective of the electrons, rather than the H+. A base must therefore have a lone pair of electrons that can take part in a bond while an acrid must have an atom that can take that alone pair of electrons. Using the reaction of HCl and h2o as an example, we use the curved pointer notation to denote how the electrons move between base and acid. Recall[4] that we use this curved arrow notation to indicate the movement of electron pair
from a source of electrons to a sink. Hither the source is the lone pair on the oxygen, and the sink is the hydrogen (which has a δ+ due to its bonding to a Cl) The 2nd pointer moves from the source (the bond between H and Cl, to the sink—the electronegative Cl which ends up with the negative charge, while the O that donated the original electron pair ends up with a positive accuse).
The Lewis model encompasses the Brønsted–Lowry model, that is, all Brønsted–Lowry acid–base reactions that can be described using the Lewis model. However, the Lewis model extends the range of reaction types that tin be considered as acid–base reactions. Take for example the reaction of ammonia (NH3) and boron trifluoride (BF3). This reaction is classified as a Lewis acid–base reaction, simply it is not a Brønsted acid–base reaction.
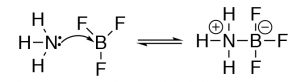
Why use different models of acrid–base chemistry? While at commencement the idea of using different models to explicate acid–base of operations chemical science may be a little confusing. Why not use the all-encompassing Lewis model for everything? It turns out that both the Brønsted–Lowry and Lewis models are particularly useful depending on the organization under consideration. The play a joke on is to recognize which is the most useful when describing, predicting, and explaining a particular type of chemical reaction.[5]
In our explorations in organic chemistry we will be using both Brønsted–Lowry (proton transfer) and Lewis (electron pair donation) models to describe acid–base of operations chemical science, depending on the type of reaction. In practice, the Brønsted–Lowry model is simple and useful; it tells you what is happening (proton transferred from acid to base) but nothing about the mechanism by which the H+ moves. For that we must turn to the Lewis model, which tells us how the electrons rearrange during the reaction. It is also important to keep in mind why these reactions happen—they are caused past an electrostatic interaction between 2 oppositely charged parts of molecules: δ – is attracted to δ + .
One farther note, all reactions are initiated past random collisions of molecules, simply only collisions that allow the electrostatic interaction of the acid and base to occur are productive (that is, collisions that involve two similarly charged parts of molecules volition not give ascent to a reaction. Again nosotros will take more to say about this after.
Acid–Base of operations Reaction Management and Position of Equilibrium
Acid base reactions begin because of electrostatic interactions, but the extent to which the reaction proceeds depends on the relative Gibbs energy of the reactants and products, that is, the overall Gibbs free energy change (ΔG) for the reaction. This is a subtle just important point: the reaction does not occur considering the products are more stable, it occurs because there is an attractive force between two reactants that have polar structures, As we will see, we can predict the relative amounts of reactants and products in a mixture (at equilibrium), based both on an understanding of molecular structures and by comparing their pKa.
Acid Strength (using the Brønsted–Lowry model): The force of an acrid, that is the degree to which information technology donates H+ to (or accepts electron pairs from) other molecules, depends on a number of factors including, obviously, the strength of the base (that is the caste to which the base donates electron pairs to other molecules) information technology reacts with. Acid and base of operations strengths are ordinarily reported using water as the solvent (i.e. as the base of operations or acid respectively), and then that acrid strengths can be compared straight. Since biological reactions take identify in aqueous solution we volition be able to extend our agreement of simple acrid base reactions to much more complex ones equally we motion forward.
The reaction for whatsoever acid HA is:
HA + H2O ⇄ H3O+ + A–
We tin can estimate the extent of the reaction (i.e., how far the reaction goes, that is the concentrations of reactants and products when the reaction reaches equilibrium) by determining the equilibrium constant Ka.
Yarda = [HthreeO+][ A–]/[ HA]
In contrast to strong inorganic acids (such as HCl, or HNO3), the equilibrium constants for many organic acids are small (ranging from 10–1 to 10-55) and it is more mutual to report pKa – which, as you volition remember, is = –log K a . A potent acrid such as HCl has a big Ka (in fact it is so large as to be meaningless) and therefore a very small (negative) pKa.
Some representative Ka and pKa values.
| Acrid | Ka | pKa |
| HCl (hydrochloric acid) | ~107 | –vii |
| CF3COOH (Trifluoroacetic acid) | 3.2 x x–1 | 0.5 |
| HF (hydrofluoric acid) | vii.2 x ten–four | 3.14 |
| CHthreeCOOH (acetic acid) | one.eight x ten–5 | 4.viii |
| H20 | 10-14 | 14 |
| CH3CH2OH (acetic acid) | 10-16 | 16 |
| NH4 + (ammonia in NH4Cl) | v.6 x 10–x | 9.25 |
| CH4 (methane) | ~10–55 | 55 |
Information technology helps to exist able to interpret these numbers in terms of the extent of the associated reaction. For case, water (which acts as both an acid and a base of operations) dissociates to a very small-scale extent. In a liter of pure water, which contains ~54 moles of water molecules (or ~54 x six.02 x x23 molecules or ~3.25 x 1025 molecules), ~10-7 moles (or ~x-7 ten 54 x half dozen.02 ten x23 molecules or ~three.25 x 1016 H3O+ ions). The weaker the acrid the college the pKa (can you explain why that is the case and what it means in terms of the relative concentrations of species at equilibrium?).
It will aid you greatly if you memorize a few important judge pK a values for mutual acids, for case alcohols tend to have a pKa of ~15, while amines accept a pKa ~33. As nosotros will meet the pKa of various carbon species is very dependent on the environment of the C-H bond, but remembering that sp3 carbon-hydrogen bonds (pKa ~55) are not likely to ionize nether any circumstances is helpful. However, it is even more important to understand the factors that affect acid strength, and be able to use them to predict and explicate the outcomes of reactions.
Another important thought to remember is that the extent of a reaction (as measured by its equilibrium constant K) is related to the alter in Gibbs energy (ΔG° = ΔH° – TΔS°) associated with that reaction. That is when we call back nigh the extent of a reaction (the concentration of reactants and products when the reaction reaches equilibrium) in terms of the relative stabilities of the reactants and products nosotros need to take into account both the enthalpy alter (ΔH°), which reflects the changes in bonding and intermolecular interactions involving both reactants and products, and the entropy modify (ΔS°) associated with the reaction system. Recall that ΔS° reflects change in the number of possible energy states and positions in the reaction organisation. For nigh organic (weak) acids, it turns out that the ΔH° of the dissociation reaction in h2o is approximately zero, considering the types of bonding and interactions that are broken and formed during the reaction are similar. Differences in ΔG for the reaction (and therefor Ka and pKa) are typically due to differences in ΔS.
- Explain why acids and bases are always (every bit pairs) found together in a system.
- What is meant by the terms conjugate acid or conjugate base?
- In the Lewis model for the HCl + water reaction, explain why you draw the arrow pointing from O to H.
- Complete these acid base of operations reactions and predict the relative amounts of reactants and products when the reaction reaches equilibrium for each reaction. Explain your predictions using your cognition of atomic and molecular structures and electronegativity.
CH3NHii + HCL ⇄
CHiiiNH2 + H20 ⇄
CH3NH– + H20 ⇄
CH3NH3 + + Htwo0 ⇄
Organic Acids and Bases
Having reviewed acids and bases using rather simple molecules (HCl and NH3), permit us move on to the more than complex world of organic acids and bases, how to identify them, how to determine relative strengths, and how to predict what will happen in any given mixture. We begin by comparing the pKa's of some organic acids. Let u.s.a. begin with ethanol (pKa ~16), a molecule that we typically do not consider to be an acrid, and acetic acid (pKa 4.8). At that place is clearly a huge deviation between the pKa's of these ii molecules, the question is can we sympathise why this is the case?
If nosotros depict out their structures we see that both have (equally expected) the acidic hydrogen bonded to the electronegative oxygen. (Make sure you lot remember why the hydrogens bonded to carbons are non as acidic as those bonded to oxygen). So why the huge difference in pKa's? To reply this question we have to remember that the extent of the reaction depends on the relative thermodynamic stability of the products—that is, the arrangement containing the conjugate base of the acid and the hydronium ion. The reactions and conjugate bases of the two are shown here (↓). 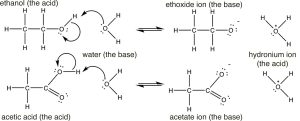 Based on their pKa values, we would predict that the ethanol dissociation reaction is rare (few ethoxide ions form) while the acerb acid dissociation reaction is more frequent. Even so annotation that even in the example of the acetic acid only about 3% of the acid molecules are dissociated in a 1M solution.
Based on their pKa values, we would predict that the ethanol dissociation reaction is rare (few ethoxide ions form) while the acerb acid dissociation reaction is more frequent. Even so annotation that even in the example of the acetic acid only about 3% of the acid molecules are dissociated in a 1M solution.
The first step in both reactions appears to exist more or less the same, an electron pair from the oxygen in water forms a bond to the electron deficient hydrogen while the O–H bond of the acrid breaks and the electrons originally associated with it the movement back to the oxygen. The deviation between the two reactions lies mainly in the mode that the negatively charged cohabit bases (ethoxide and acetate) behave, and the way that they are solvated by the solvent (h2o). For ethoxide (ethanol's conjugate base of operations), the actress negative charge is localized onto the oxygen, which leads to a concentration of charge. H2o molecules are strongly attracted to the ethoxide anion, an interaction that limits the mobility of the Resonance Structures Resonance Hybrid water molecules and results in a decrease in entropy (ΔS is negative). In contrast, in acetate (acetic acrid'due south conjugate base), the negative accuse is delocalized onto both oxygens (fifty-fifty though information technology is often drawn as if it was associated with 1 only not the other). Nosotros can illustrate this in 2 ways (or more than!) by drawing arrows to point how the extra electron pair can movement from 1 oxygen to the other; it looks similar this (→).
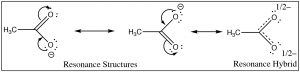 The actual structure has a partial negative accuse on both oxygens. This pair of structures is often referred to as a resonance structure and the procedure is termed resonance but the proper noun is misleading. In fact the actual structure, the resonance hybrid, does NOT involve the electrons (and the double bond) moving back and forth between the two oxygen atoms. Past a biological (and non completely sensical) analogy nosotros might say it is a mule or a hinny—the offspring of a cross betwixt a equus caballus and a donkey.[6] Simply equally a mule (or a hinny) is not bouncing back and forth between being a horse and beingness a ass, so the resonance hybrid actually exists as a new species[vii], with an actual structure that is partway between the 2 (drawn) resonance structures. In this case, nosotros are using two bonding models (a valence bond and a delocalized molecular orbital model) to describe the construction of acetate anion. The localized valence bond model involves a sigma single bond framework that connects the atoms and provides the molecular shape. The delocalized molecular orbital model describes a pi bond that connects both Os to the C. We tin visualize the anion as a planar sp2 hybridized carbon continued to a methyl group and ii oxygens by sigma bonds together with a three atom 2 electron pi bond that extends over the O–C–O framework (→). The effect is that in the acetate ion the negative accuse is delocalized over two oxygens, rather than beingness concentrated on only i atom every bit it is in the
The actual structure has a partial negative accuse on both oxygens. This pair of structures is often referred to as a resonance structure and the procedure is termed resonance but the proper noun is misleading. In fact the actual structure, the resonance hybrid, does NOT involve the electrons (and the double bond) moving back and forth between the two oxygen atoms. Past a biological (and non completely sensical) analogy nosotros might say it is a mule or a hinny—the offspring of a cross betwixt a equus caballus and a donkey.[6] Simply equally a mule (or a hinny) is not bouncing back and forth between being a horse and beingness a ass, so the resonance hybrid actually exists as a new species[vii], with an actual structure that is partway between the 2 (drawn) resonance structures. In this case, nosotros are using two bonding models (a valence bond and a delocalized molecular orbital model) to describe the construction of acetate anion. The localized valence bond model involves a sigma single bond framework that connects the atoms and provides the molecular shape. The delocalized molecular orbital model describes a pi bond that connects both Os to the C. We tin visualize the anion as a planar sp2 hybridized carbon continued to a methyl group and ii oxygens by sigma bonds together with a three atom 2 electron pi bond that extends over the O–C–O framework (→). The effect is that in the acetate ion the negative accuse is delocalized over two oxygens, rather than beingness concentrated on only i atom every bit it is in the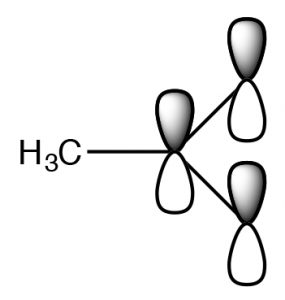 ethoxide ion. The effect is that the interactions of the acetate with solvent water molecules is not as strong, so that the water molecules are non as ordered, pregnant that the water is not as ordered around the molecule and the entropy alter is not every bit negative. The effects of delocalizing charge over more than than i atom play a major role in predicting the outcomes of a wide range of reactions. We note that ΔS is notwithstanding negative since the creation of a charged species however leads to increased ordering of solvent molecules.
ethoxide ion. The effect is that the interactions of the acetate with solvent water molecules is not as strong, so that the water molecules are non as ordered, pregnant that the water is not as ordered around the molecule and the entropy alter is not every bit negative. The effects of delocalizing charge over more than than i atom play a major role in predicting the outcomes of a wide range of reactions. We note that ΔS is notwithstanding negative since the creation of a charged species however leads to increased ordering of solvent molecules.
I manner to predict whether charge can be delocalized is to determine whether resonance structures can be drawn for the charged species. For example: try disarming yourself that you cannot describe resonance structures for ethanol.
Resonance is not the only fashion to stabilize charge. Typically, resonance occurs through aconjugated pi bail system, such equally occurs inside the –COtwo – part of an organic acid, simply how do we business relationship for the deviation in the acidities of acetic acid (pKa 4.eight) and trifluoroacetic acid (→) (pKa 0.v), even though they both have the carboxylate functional grouping? The deviation between the ii lies in the fact that the charge on the trifluoroacetate anion is delocalized by two distinct mechanisms. As in acetate, the negative accuse is delocalized by resonance 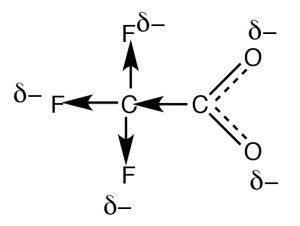 through the pi bonding system; in addition it is also delocalized onto the fluorines by the fact that the highly electronegative fluorine atoms (more than electronegative than O) withdraw electrons from the methyl carbon through the sigma bonds, which in turn withdraws electrons from the next carbon, and in turn from the two oxygens (a process known as "induction"). The result is that the negative charge is "smeared out" over even more atoms, making the anion even less likely to cause a solvent molecule ordering (reducing the upshot on ΔS). As you might expect, the inductive effect is altitude dependent (perhaps yous can predict the upshot of adding more CHii groups between the CF3 and COtwo groups).
through the pi bonding system; in addition it is also delocalized onto the fluorines by the fact that the highly electronegative fluorine atoms (more than electronegative than O) withdraw electrons from the methyl carbon through the sigma bonds, which in turn withdraws electrons from the next carbon, and in turn from the two oxygens (a process known as "induction"). The result is that the negative charge is "smeared out" over even more atoms, making the anion even less likely to cause a solvent molecule ordering (reducing the upshot on ΔS). As you might expect, the inductive effect is altitude dependent (perhaps yous can predict the upshot of adding more CHii groups between the CF3 and COtwo groups).
- Using resonance structures predict which is more acidic: Chalf dozenHvOH or CH3CH2OH?
- Describe structures to evidence how sodium ethoxide and sodium acetate are solvated in water, and use them to show why the negative entropy modify for the germination of sodium acetate is smaller than that of sodium ethoxide.
- Consider the pKa'south of the iii chlorobutanoic acids: CHthreeCH2CHClCOOH (pKa 2.86), CHthreeCHClCHiiCOOH (pKa 4.05), and CHtwoClCHtwoCHtwoCOOH (pKa 4.53). Describe structures and employ them to explicate why these carboxylic acids have different pKa's.
Organic Bases
Every bit noted previously, there are no acids without bases, and vice versa. Even if we are but discussing H+ (proton) transfer, it is (arguably) easier to recall about the base of operations using a Lewis model. That is, a base has an electron pair available for donation into a bond with the acid. Recall that almost everything that has a pair of non-bonding electrons (sometimes chosen a lone pair) can act as a base. The well-nigh common types of organic bases ofttimes have a nitrogen cantlet somewhere in their construction. If we compare the basicity of N, O and F, each of which accept lone pairs that are could potentially exist donated, nitrogen is the to the lowest degree electronegative and therefore the all-time able to donate its electrons into a bond, since its alone pair is least attracted past the nucleus. Fluorine, the about electronegative chemical element, holds its electrons very close to the nucleus, and under normal circumstances would non be considered equally a base of operations.
Oxygen, since it is more electronegative than nitrogen is not every bit stiff a base of operations, therefore when ammonia and water are mixed, the only reaction that occurs (and that to a relatively small extent) is a proton transfer from h2o to ammonia.
NH3 + H2O ⇆ NH4 + + –OH
The equilibrium constant for this reaction is 1.viii 10 ten–5 (most of the species in the mixture at equilibrium are reactants)
*insert prototype here*
Here are some organic bases (→). Note that they are components of a wide range of biologically active molecules, including Deoxyribonucleic acid, hormones and pharmaceuticals. Equally we volition see the basic nitrogen provides an important way to understand the reactivity of a item species.
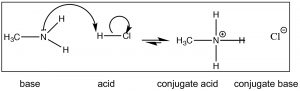 For now, however, let us get-go with a simpler base such as methylamine (CHiiiNH2) the simplest nitrogenous organic base. Methylamine reacts with acids (↓) in much the same way that ammonia does; information technology will react with a strong acid like HCl(aq) to produce methylammonium chloride.
For now, however, let us get-go with a simpler base such as methylamine (CHiiiNH2) the simplest nitrogenous organic base. Methylamine reacts with acids (↓) in much the same way that ammonia does; information technology will react with a strong acid like HCl(aq) to produce methylammonium chloride.
Call up that the position of equilibrium can be predicted by comparison the strength (pKa's) of the two acids. HCl (pKa –vii) is a much stronger acid than CH3NHthree + (pKa ~10) and therefore we predict that the equilibrium of the methylamine + HCl reaction will lie well to the right. Now consider the reaction in which methylamine reacts with acetic acid (↓).
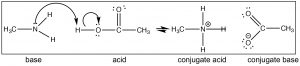
Again we tin predict the position of equilibrium by comparing pKa's of the conjugate acids (acetic acrid 4.8 and CH3NH3 + ~ 10). Notice that you tin predict the structure of the products simply past post-obit the flow of electrons. We could alter the CH3 (methyl) groups on either methylamine and acetic acid to a broad range of different groups and still exist able to predict the production easily, as long as you lot recognize that the reaction that takes place is a (simple) proton transfer (acrid–base). For example, look at the construction of cocaine (above): can y'all predict what will happen if information technology were reacted with acerb acid? What would be the structure of the production?
Molecules that comprise both an acid and a base:
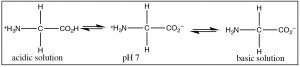
The most common example of a molecule that act as both an acrid and a base is of grade water considering it has both a potentially acidic hydroged, and a solitary pair that can accept the proton. However, since this is organic chemistry, where h2o is not as common a solvent, allow us consider the grade of molecules that have both acidic and bones domains simultaneously. The most biologically of import such molecules are the amino acids, which have both an amino group and a carboxylic acid. A subset of the possible amino acids are those used in biological systems to assemble polypeptides. Amino acids (or rather the α-amino acids) contain both a carboxylic acid and an amino group fastened to a central carbon (the α-carbon). The generic structure is given here (→) where R stands for a broad range of side bondage.[viii] 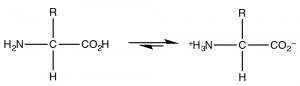 At pH seven the amino acrid exists in what is know equally a zwitterionic form, in which the carboxylic acid group is negatively charged while the amino group is positively charged. At no time would an amino acrid (dissolved in water) exist in an un-ionized form. Nosotros can predict what class would be nowadays at different pH's by because the pKa's of the species involved.
At pH seven the amino acrid exists in what is know equally a zwitterionic form, in which the carboxylic acid group is negatively charged while the amino group is positively charged. At no time would an amino acrid (dissolved in water) exist in an un-ionized form. Nosotros can predict what class would be nowadays at different pH's by because the pKa's of the species involved.
Effect of pH on acrid base of operations reactions
So far, nosotros take discussed situations when the acid or base of operations is added to solution of pure water. Pure water has a pH of seven, and [HiiiO+] = [–OH]. Now let us consider what happens when we change the pH of the solution. For instance consider a situation in which we dissolve a simple organic acid (CH3CO2H, acerb acid) in a solution that has a pH > 7, that is where the [–OH] > [HthreeO+]; nether these atmospheric condition the extent of the acerb acid's ionization is increased. 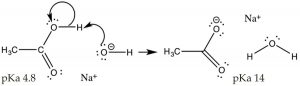 Remember that in 1M acetic acid only ~3% of the acrid is ionized at pH 7. If we change the solution to make it bones by adding NaOH, the excess of stiff base (–OH) volition completely deprotonate the acid. At equilibrium, the reaction will now favor products over reactants (i.eastward. information technology will move to the right). What nosotros have done here is drive the acerb acid ⇆ acetate reaction to the right, increasing the concentration of acetate, which is an application of Le Châtelier'southward principle). Note that Na+, derived from the addition of the NaOH used to adjust the pH, is present but does not take part in the reaction – for this reason information technology referred to as a "spectator ion". Another, perhaps simpler, way to predict the issue of this reaction is to utilise the pKa values of the two acids (CH3CO2H, 4.8 and H2O, 14), clearly acetic acid is a much stronger acid than water, and therefore the equilibrium position for this reaction will lie over to the right in favor of the weakest acid and the weakest base of operations. What we have done here is alter the acetic acid, which is a polar organic molecule, into acetate, an ionic species.
Remember that in 1M acetic acid only ~3% of the acrid is ionized at pH 7. If we change the solution to make it bones by adding NaOH, the excess of stiff base (–OH) volition completely deprotonate the acid. At equilibrium, the reaction will now favor products over reactants (i.eastward. information technology will move to the right). What nosotros have done here is drive the acerb acid ⇆ acetate reaction to the right, increasing the concentration of acetate, which is an application of Le Châtelier'southward principle). Note that Na+, derived from the addition of the NaOH used to adjust the pH, is present but does not take part in the reaction – for this reason information technology referred to as a "spectator ion". Another, perhaps simpler, way to predict the issue of this reaction is to utilise the pKa values of the two acids (CH3CO2H, 4.8 and H2O, 14), clearly acetic acid is a much stronger acid than water, and therefore the equilibrium position for this reaction will lie over to the right in favor of the weakest acid and the weakest base of operations. What we have done here is alter the acetic acid, which is a polar organic molecule, into acetate, an ionic species.
Acetic acid is a modest organic molecule; since information technology is polar information technology can interact with water (though intermolecular forces), therefore acetic acid is very soluble in water (indeed it is miscible with water (information technology has unlimited solubility.)[9] But now, let us consider the effect of increasing the length of the hydrocarbon group of the organic acid on its molecular backdrop. Acetic acid has a methyl (CHiii–) group, the smallest possible hydrocarbon. In contrast dodecanoic (lauric) acid has a 12-carbon hydrocarbon chain (CHthree[CH2]xi–) and has a solubility in water of 0.063 g/L (~xxx mM) at 25 °C, which is much less that of acetic acrid.[x] Every bit the hydrocarbon (non-polar) part of the molecule increases in length, solubility in water decreases: the ΔG of the procedure of dissolving the organic acrid in water becomes more than positive. This subtract in solubility is primarily due to a negative entropy change (ΔS) acquired by the cocky-organization of water molecules around the hydrocarbon "tail" of the molecule.  Now let united states of america consider the behavior of ionized sodium dodecanoate (the sodium salt of dodecanoic acrid); it, like many ionic species, information technology is soluble in h2o. Although as you may call up, this is a different form solubility – the soluble species is not isolated molecules but rather molecular complexes known as micelles (→).[11] The issue of this is we can "solubilize" organic acids in h2o past deprotonating them, but if nosotros then lower the pH, the organic acid volition separate from solution again.
Now let united states of america consider the behavior of ionized sodium dodecanoate (the sodium salt of dodecanoic acrid); it, like many ionic species, information technology is soluble in h2o. Although as you may call up, this is a different form solubility – the soluble species is not isolated molecules but rather molecular complexes known as micelles (→).[11] The issue of this is we can "solubilize" organic acids in h2o past deprotonating them, but if nosotros then lower the pH, the organic acid volition separate from solution again.
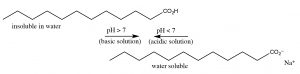
Organic bases can be solubilized in a like mode, except that now the solution must exist made acidic. For example, a nitrogenous base of operations with a large not-polar grouping such as dodecylamine (C12H27N) has a solubility of about three.5 g/L (~20mM), only at acidic pHs it is completely soluble. Contrast the solubility of dodecyl amine with cadaverine (NHiiCHtwoCH2CH2CHiiCHtwoNH2), the compound that smells like its proper noun, which is completely miscible with h2o because it has 2 polar amino groups. It turns out that we tin predict the pH at which a detail acid or base of operations protonates or deprotonates. You may recall from general chemistry that the pH of weak acids and their conjugate bases (like nearly organic species) can be described using the Henderson Hasselbalch equation (→).
One way to work with this equation is to notation that [acid] = [conjugatepH =pKa + log [latex]\frac{[base]}{[acid]}[/latex] base] when the pH of the system is equal to pKa of the acid. At pH'south below pKa, [acrid] > [conjugate base of operations], there is more acrid than base, and vice versa for pH > pKa. Therefore, past adjusting the pH nosotros can change the concentrations of conjugate acid and base to suit our purposes, or nosotros can predict the relative concentrations at whatsoever pH. For example, acetic acid with a pKa of 4.eight would have 50% CH3CO2H, and 50% CH3COii –Na+, at a pH of 4.8. If the pH falls below 4.8 the concentration of protonated acid will increase, and if it rises the concentration of acetate ion will increase.
This power to transform an organic substance from an insoluble (in water) molecule to a soluble ionic species can be very useful. I mutual example stems from the fact that many pharmaceutical drugs are organic substances that are insoluble in aqueous solutions (like cytoplasm or claret). If these substances were introduced into the body in their not-ionized form they would not dissolve, and therefore be inactive. If yous check the labels on many prescription bottles you lot will meet that the drug is administered as a table salt. Consider norepinephrine (→), a hormone that is often administered intravenously to counteract the effects of allergic reactions. 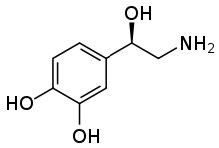 It is administered equally a salt of tartaric acid to ensure that it is soluble in the blood stream.
It is administered equally a salt of tartaric acid to ensure that it is soluble in the blood stream.
You lot may come up across another example of this phenomenon (that acids are soluble in basic solution, and bases are soluble in acid solution) if you take the organic chemistry laboratory course. If your product has an acidic or basic moiety in its structure, y'all can extract the substance into aqueous (acid or bones) solution, washing away all the organic by-products with an organic solvent, and then regenerating the acidic or basic substance. This is an important purification method for many substances, because it allows the compound of interest to be separated into aqueous solution and then regenerated just past adding or subtracting a proton.
 When we consider biomolecules (that is, organic molecules found in organisms) the situation is non so articulate cut; most biomolecules have a variety of acidic and basic groups every bit function of their structure. Even the simplest amino acid, glycine (→) exist in a variety of protonated and deprotonated forms depending on the pH.
When we consider biomolecules (that is, organic molecules found in organisms) the situation is non so articulate cut; most biomolecules have a variety of acidic and basic groups every bit function of their structure. Even the simplest amino acid, glycine (→) exist in a variety of protonated and deprotonated forms depending on the pH.
Ane thing that becomes articulate is that individual amino acids are e'er charged regardless of the pH, then they are water-soluble. But the extent of the protonation/deprotonation reactions is pH dependent. As we will come across this has a number of ramifications for a wide range of biological molecules, because they will comport very differently in unlike pH solutions. This is i important reason why most biological systems are buffered so that they remain at a fairly abiding pH.
- If you take a mixture of benzoic acid C6H5CO2H (pKa iv.ii), toluene, Chalf-dozenHvCHthree and aniline hydrochloride (pKa of CsixH5NH3 + 4.6). Which substance will exist soluble in aqueous acidic solution, which will be soluble in aqueous basic solution, which volition not exist soluble in water?
- Outline a scheme for separating these 3 substances by using their differing solubilities in organic and aqueous solutions of different pHs.
Lewis Acids and Bases, Electrophiles and Nucleophiles
Equally we accept seen, any reaction in which a proton (H+) is transferred from one molecule to another can exist considered as a Lewis acrid–base reaction, merely now it is fourth dimension to broaden the scope of Lewis acid–base of operations reactions. The structural requirement for a Lewis base is essentially the aforementioned as those we discussed for a Brønsted base. That is, a Lewis base must have an accessible lone pair of electrons that can exist donated into a bail with a Lewis acrid. 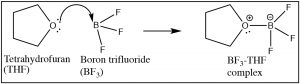 For example, many (but not all) nitrogen and oxygen containing molecules accept such available lone electron pairs and so can be considered every bit Lewis bases.[12] It is the Lewis acrid that tin take a number of dissimilar forms (and so, can be harder to recognize). A Lewis acid must exist able to accept a pair of electrons. In practice this means a multifariousness of substances (besides H+) tin can act as Lewis acids: for example, whatever species with empty orbitals that are energetically attainable can be a Lewis acid.
For example, many (but not all) nitrogen and oxygen containing molecules accept such available lone electron pairs and so can be considered every bit Lewis bases.[12] It is the Lewis acrid that tin take a number of dissimilar forms (and so, can be harder to recognize). A Lewis acid must exist able to accept a pair of electrons. In practice this means a multifariousness of substances (besides H+) tin can act as Lewis acids: for example, whatever species with empty orbitals that are energetically attainable can be a Lewis acid. 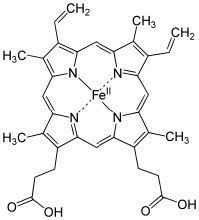 Mutual examples of this situation are compounds of Group III elements (specifically B and Al); these have only iii valence electrons. Examples include BF3 (↑) and AlCl3,[13] both of which accept a partial positive accuse on the central atom and an empty orbital that tin accept electrons. Other examples of Lewis acids are metal ions such as Fe2+, Atomic number 263+, Cu2+, and Mg2+; these, by definition, have empty orbitals. The same situation holds true for many transition metallic salts, for case TiClfour and NiCl2.[14] In biological systems, examples of Lewis acid–base complexes include the active site of the oxygen ship circuitous in hemoglobin (and myoglobin), which consists of an atomic number 26 ion complexed with 4 nitrogens, which are function of a porphyrin band. A similar iron-porphyrin complex is constitute associated with the cytochrome proteins that participate in the ATP synthesis reaction associated with in the electron transport concatenation of the mitochondria (→). Chlorophyll, the green pigment that is part of the light capture organisation in algae and plants has a like structure, except that the Lewis acrid at the center of the complex is Mg2+ rather than Iron2+. This has the interesting result of making chlorophyll species appear to be greenish, rather than the red observed in blood. This is acquired by the difference free energy gaps between the molecular orbitals in an Fe complex as compared to a Mg complex with a porphyrin band. Nosotros will hash out this upshot in more item later. As we will also see later, Lewis acids are important form of reagents in organic chemistry considering they tin interact with a broad range of bases.
Mutual examples of this situation are compounds of Group III elements (specifically B and Al); these have only iii valence electrons. Examples include BF3 (↑) and AlCl3,[13] both of which accept a partial positive accuse on the central atom and an empty orbital that tin accept electrons. Other examples of Lewis acids are metal ions such as Fe2+, Atomic number 263+, Cu2+, and Mg2+; these, by definition, have empty orbitals. The same situation holds true for many transition metallic salts, for case TiClfour and NiCl2.[14] In biological systems, examples of Lewis acid–base complexes include the active site of the oxygen ship circuitous in hemoglobin (and myoglobin), which consists of an atomic number 26 ion complexed with 4 nitrogens, which are function of a porphyrin band. A similar iron-porphyrin complex is constitute associated with the cytochrome proteins that participate in the ATP synthesis reaction associated with in the electron transport concatenation of the mitochondria (→). Chlorophyll, the green pigment that is part of the light capture organisation in algae and plants has a like structure, except that the Lewis acrid at the center of the complex is Mg2+ rather than Iron2+. This has the interesting result of making chlorophyll species appear to be greenish, rather than the red observed in blood. This is acquired by the difference free energy gaps between the molecular orbitals in an Fe complex as compared to a Mg complex with a porphyrin band. Nosotros will hash out this upshot in more item later. As we will also see later, Lewis acids are important form of reagents in organic chemistry considering they tin interact with a broad range of bases.
Electrophiles and Nucleophiles
The next logical step in expanding our ideas about Lewis acids and bases is to consider reactions that involve carbon. We will first consider reactions in which carbon acts like the Lewis acid, that is, it accepts a pair of electrons to form a new bond with a Lewis base. So, what situations would nosotros brand a carbon act in this way? Nosotros can dominion out (for at present) carbon compounds with an empty orbital (akin to boron). Why? Considering all stable carbon compounds form iv bonds and there are no low-lying empty orbitals that can exist used to accept electrons.
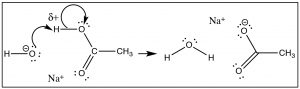 But let us outset wait at the proton (H+) transfer reaction as a model (→). In this case the bond with the Lewis base (OH-) is formed at the same time as the bond to the cohabit base (of the acrid) is broken. We encounter that the δ+ on the H means that the bail to the H is partially ionized. The H is "on the way" to becoming H+—a species that does have an empty and accessible orbital. The δ+ on the H attracts the negative (or δ–) charge on the base of operations, and the reaction is initiated, forming a new bail between the O and the H, and at the same fourth dimension breaking the erstwhile O–H bail.
But let us outset wait at the proton (H+) transfer reaction as a model (→). In this case the bond with the Lewis base (OH-) is formed at the same time as the bond to the cohabit base (of the acrid) is broken. We encounter that the δ+ on the H means that the bail to the H is partially ionized. The H is "on the way" to becoming H+—a species that does have an empty and accessible orbital. The δ+ on the H attracts the negative (or δ–) charge on the base of operations, and the reaction is initiated, forming a new bail between the O and the H, and at the same fourth dimension breaking the erstwhile O–H bail.
We tin imagine that a carbon compound with a δ+ on the C might deport in a very similar manner. 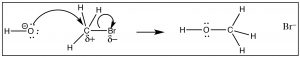 In this molecule (HthreeCBr) the C-Br bond is polarized so that at that place is a small positive charge on the C, which attracts the negatively charged hydroxide (→). Formation of the O–C bail occurs with the simultaneous breaking of the C-Br bail.
In this molecule (HthreeCBr) the C-Br bond is polarized so that at that place is a small positive charge on the C, which attracts the negatively charged hydroxide (→). Formation of the O–C bail occurs with the simultaneous breaking of the C-Br bail.
Consider the analogies betwixt these two reactions – the mechanisms of how and why the electrons move are like. The only real deviation between the two reactions is that in the offset an H+ is transferred from an O (on the carboxylic acrid) to the OH–, while in the 2nd, a methyl grouping is transferred to the OH–. Now for a change in nomenclature: when such a reaction involves a C atom (a carbon center) rather than call the electron deficient carbon a Lewis acid, we call it an electrophile (electron or negative charge loving). Similarly, the hydroxide ion (which acts as a Lewis base of operations) is now called a nucleophile (positive charge loving). This change in terminology is not but to confuse students! In fact, in that location are subtle differences between Lewis acids and bases and electrophiles and nucleophiles that brand the stardom betwixt the 2 useful. In particular, while all Lewis bases are nucleophiles, as we will see, not all nucleophiles are bases.
So now nosotros have to ask ourselves, what factors make a particular C within a molecule an electrophile? How tin can we recognize a nucleophile? What criteria exercise we use to estimate the force of a item electrophile or an nucleophile? Can we ever get carbon to act equally a nucleophile? If we can answer these questions, we tin can predict the outcome of a broad array of reactions.
What makes a particular carbon an electrophile?
The simplest of organic compounds are hydrocarbons, and the simplest of hydrocarbons are known as alkanes. Alkanes typically have the formula CnH2n+2 (or if CnorthH2n if there is ane ring of carbons, subtract 2H for every extra ring). All of the bonds within an alkane are sigma (unmarried) bonds; they practise non contain pi (double) bonds.[xv] In an paraffin, each carbon is fully saturated, it makes four unmarried bonds and (every bit noted above) there are no double or triple bonds. C-C bonds are of class, completely non-polar since the electrons are equally distributed between 2 identical atoms, however C-H bonds are also relatively not-polar since the electronegativities of C and H are quite like. In practice this means that alkanes are limited in their reactivity. The most common reactions that an alkane can take office in are reactions with oxygen to produce CO2 and HtwoO. This reaction is highly exothermic, although there is a significant activation energy, then it requires an initial input of free energy (typically a spark, a burning lucifer) to start the reaction, but then the free energy from the formation of the strong C=O and O-H bonds (which is why the reaction is exothermic) can be used to initiate more reaction. The actual reaction mechanism is circuitous; it proceeds via a series of highly reactive (unstable) free radicals (species with unpaired electrons)[16]. While this reaction is obviously highly important—this is even so how we generate much of the free energy to run our cars and electrical power stations, from an organic chemical science perspective it is non very interesting in large part because it is more than or less uncontrollable. That is, if you have enough oxygen once started the reaction generates COii and H2O, regardless of which hydrocarbon you begin with. [17]
All this is to say that alkanes are not good candidates for the kinds of reactions we are considering, they have neither nucleophile nor electrophilic carbons. So, let the states plow our attention to carbon compounds with elements other than C and H and both sigma and pi bonds (this is, of course, the rest of organic chemistry). Here we detect a very different situation: the range of reactions and the types of products tin can seem almost unlimited. While it is incommunicable (and certainly undesirable) to memorize every reaction and every potential product, it is possible to organize your understanding of chemic systems and then that you can make plausible predictions as to which reactions may occur. By knowing reaction mechanisms, and when they are relevant, you can also predict which reactions will occur and therefore what products will course. As you might recognize, this is the same strategy nosotros have used to consider acid–base of operations reactions, which can exist understood much more broadly than elementary proton (H+) transfer reactions. Thinking in an electrophile-nucleophile context provides an entrée into much of organic chemistry.
For reactions (other than reactions involving free radicals, like combustion) to occur, in that location is generally a "handle" inside the substrate: a place where the electron density is non evenly distributed, a site at which reactants of opposite accuse collaborate (and react). In the instance nosotros used previously, the electrophilic carbon has a δ+ on it; this fractional charge arose because the C was bonded to a more electronegative element. Such a partially positively charged C is attractive to any species with a negative (or fractional negative) charge. 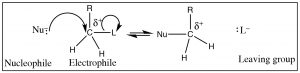 Annotation that, for now, we are going to restrict the blazon of carbon atom that we are considering to either a primary (that is a carbon with only one alkyl group (denoted by R) and 2 hydrogens, CH2R–) or a methyl carbon (CHiii–). Every bit nosotros volition see things get more complicated when we starting time to add more than alkyl groups effectually the site of attack—and so we will come back to that later on.
Annotation that, for now, we are going to restrict the blazon of carbon atom that we are considering to either a primary (that is a carbon with only one alkyl group (denoted by R) and 2 hydrogens, CH2R–) or a methyl carbon (CHiii–). Every bit nosotros volition see things get more complicated when we starting time to add more than alkyl groups effectually the site of attack—and so we will come back to that later on.
To identify such a partially positively charged C i would look for C's bonded to groups (atoms) that are more electronegative, that is, that volition act to withdraw electrons from the carbon (denoted past L below). But since carbon cannot form more than than 4 bonds as the nucleophile comes in and forms a bond, another bail must interruption. The electronegative atom (L) (or group of atoms), is known as the "leaving group" (oh, how dull) needs to be stable when it leaves with the extra pair of electrons. We can, in fact, predict the characteristics of a practiced leaving group. For instance, the bail to the leaving group should be polarized, and since the leaving group takes the electron pair with it, the group should be stable with this extra pair of electrons on it (Fifty–). Another fashion of saying this is that the leaving group should be electronegative and breaking the C-L bail should produce a weak base. Halide ions are examples of good leaving groups, and their club of reactivity is I– > Br– > Cl– > F–. This ranking mirrors their acid strength rankings—that is, Hi is the strongest acid and HF is the weakest—which means F– is the strongest base (and therefore least likely to exit)
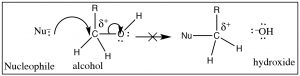 So, what about oxygen, in the grade of an booze O–H group, equally a leaving group? (→) It certainly fulfills the requirement that the C–O bail be polarized, but if you follow the reaction through it would mean that the leaving group would exist a hydroxide ion (– OH), a very strong base. Therefore, alcohols (ROH) are not likely to exist attacked by a nucleophile.
So, what about oxygen, in the grade of an booze O–H group, equally a leaving group? (→) It certainly fulfills the requirement that the C–O bail be polarized, but if you follow the reaction through it would mean that the leaving group would exist a hydroxide ion (– OH), a very strong base. Therefore, alcohols (ROH) are not likely to exist attacked by a nucleophile.
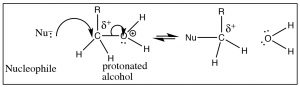 There are means, however, ways to make an alcohol reactive. For example, if we tin can conduct out the reaction in an acidic solution, the alcohol will exist protonated (at least some of the time), and therefore the leaving group will be a h2o molecule, a stable entity (→).
There are means, however, ways to make an alcohol reactive. For example, if we tin can conduct out the reaction in an acidic solution, the alcohol will exist protonated (at least some of the time), and therefore the leaving group will be a h2o molecule, a stable entity (→).
What makes a practiced nucleophile?
As nosotros accept noted, a Lewis base is also a nucleophile, so the trends you lot have learned about the strengths of Lewis bases too hold for nucleophiles. So, for example, nucleophilicity decreases beyond a row in the periodic table so NHthree > H2O > HF in the same way as base strength does (recall this is because the lone pair is more available on the Due north than on F). Only since this is organic chemistry, we should have some organic groups dangling off the nucleophiles. 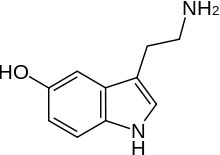 So for example, instead of a hydroxide nucleophile, nosotros could apply an alkoxide nucleophile (for example, CH3CH2O– Na+ sodium ethoxide), or we could utilize amine nucleophiles like serotonin (the nitrogen in the NH2 group here is more nucleophilic than the OH group, and the N in the band). In addition, if we compare nucleophiles with the same nucleophilic cantlet, a negatively charged species is more nucleophilic than the uncharged form, then OH– > HiiO, and NH2– > NH3 (and by analogy any organic derivatives behave the same way).
So for example, instead of a hydroxide nucleophile, nosotros could apply an alkoxide nucleophile (for example, CH3CH2O– Na+ sodium ethoxide), or we could utilize amine nucleophiles like serotonin (the nitrogen in the NH2 group here is more nucleophilic than the OH group, and the N in the band). In addition, if we compare nucleophiles with the same nucleophilic cantlet, a negatively charged species is more nucleophilic than the uncharged form, then OH– > HiiO, and NH2– > NH3 (and by analogy any organic derivatives behave the same way).
Besides the nucleophiles that are easily recognizable because they are bases, at that place is another course of nucleophiles that are somewhat different; they have a alone pair of electrons, just they are not particularly basic. The nearly common examples are the halide ions, which are weak bases and expert leaving groups. And so, the question arises: why are halide ions such skilful nucleophiles? The reason for this has to practise with their polarizability (that is, the extent to which an electron cloud can get distorted) of the nucleophile. A very large anion-like iodide has a very polarizable electron cloud because the electrons extend much further out from the nucleus than, for instance, the electron cloud in fluoride. This means that the electron cloud for iodide can begin partial bond formation to the carbon much earlier than the one for fluoride, and therefore iodide reacts much faster than fluoride.[xviii] This logic allows us to explain why the nucleophilicity of halide ions increases equally you go down a grouping: I– > Br– > Cl– > F–.
Although we will return to this reaction in greater detail later, let us have a wait at the range of possible reactions that this generic scheme enables the states to predict – with the caveat that we are considering elementary carbon substrates. Reactions like this are chosen nucleophilic substitutions, because the species that attacks the carbon is a nucleophile, and the overall issue of the reaction is that nosotros take substituted the nucleophile for the leaving group. This item example is called an S N ii reaction which stands for Substitution, Nucleophilic, 2 nd Club, and nosotros will come back to discuss the reaction in much more than particular later.
Another type of carbon nucleophile
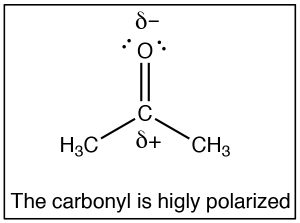 The SouthN2 reaction is a mainstay of organic chemistry, by varying the substrate (carbon electrophile) the leaving grouping, and the nucleophile we can construct a huge array of different compounds. Some other very important type of compound that has an electrophilic carbon (i.e. a carbon that is subject to nucleophilic attack) is ane which contains a carbonyl grouping (C=O). The carbonyl group is highly polarized, with a large δ+ on the carbon. This tin can be rationalized past the idea that there are 2 bonds to the electronegative oxygen and therefore the oxygen has even more tendency to pull electrons away from the carbon than a single bonded oxygen would.
The SouthN2 reaction is a mainstay of organic chemistry, by varying the substrate (carbon electrophile) the leaving grouping, and the nucleophile we can construct a huge array of different compounds. Some other very important type of compound that has an electrophilic carbon (i.e. a carbon that is subject to nucleophilic attack) is ane which contains a carbonyl grouping (C=O). The carbonyl group is highly polarized, with a large δ+ on the carbon. This tin can be rationalized past the idea that there are 2 bonds to the electronegative oxygen and therefore the oxygen has even more tendency to pull electrons away from the carbon than a single bonded oxygen would. 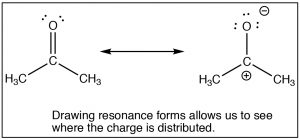 1 manner to visualize this is to draw resonance structures for the carbonyl group equally shown, where the electrons from the double bond are now located on the O. We will come back to how to draw resonance forms in much more item later on.
1 manner to visualize this is to draw resonance structures for the carbonyl group equally shown, where the electrons from the double bond are now located on the O. We will come back to how to draw resonance forms in much more item later on.
Once we understand how compounds with carbonyl groups are polarized, nosotros tin predict (at to the lowest degree for the first step) how these compounds will react. For case, if we have a reasonably proficient nucleophile (here shown every bit Nu–)we might predict that it would attack at the carbonyl carbon. 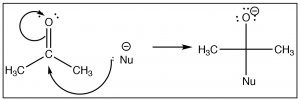 The difference in this reaction and an SNtwo reaction is that there is no leaving group. Instead the electrons from ane of the C-O bonds move onto the oxygen as shown.
The difference in this reaction and an SNtwo reaction is that there is no leaving group. Instead the electrons from ane of the C-O bonds move onto the oxygen as shown.
There are a number of ways that this reaction tin proceed, the most obvious of which is that if the reaction is in contact with a solvent that has acidic protons (e.g. h2o or an booze), the O– tin only protonate in an acid base reaction. As we will see later, the form of the reaction also depends on what the nucleophile is. Here nosotros will requite the simplest instance which is the reaction of a ketone (acetone) with a carbon nucleophile (CHiiiCH2Li, ethyl lithium). For now, nosotros volition not worry about how to brand ethyl lithium, but balance bodacious it is possible! When the negatively charged carbon electrophile adds to the carbonyl nosotros brand a new carbon-carbon bond. This is followed by addition of water to protonate the oxygen, to produce an alcohol. The overall reaction is a nucleophilic addition.
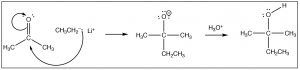
Effort your paw at predicting the outcomes for these reactions past drawing pointer pushing mechanisms.
- CHiiiCH2I + NaOH →
- CH3Br + NaN3 →
- CHiiiCHiiCl + NH2CHthree →
- CHiiiOH + H+ →
What nucleophile and electrophile would you react together to form these products?
- CH3OH + Br–
- CHthreeCH3NH3 + + Cl–
- Construct a generalizable model for the SouthDue north2 reaction and explain the role of the substrate (the carbon electrophile), the leaving grouping, and the nucleophile.
Construct a generalizable model for the nucleophilic addition reaction and explain the role of the substrate (the carbon electrophile), and the nucleophile. What functional groups would undergo a nucleophilic addition? - What would make a carbon in a compound a nucleophile? How could you get about making a detail carbon nucleophilic?

Products Of Acid Base Reaction,
Source: https://openbooks.lib.msu.edu/oclue/chapter/chapter-1-acid-base-reactions/
Posted by: walkerdeboyfaing.blogspot.com
0 Response to "Products Of Acid Base Reaction"
Post a Comment